

Computational Chemistry and Molecular Modeling
The enormous increase in computational power of common CPUs, the rise of scientific computing on GPUs (graphic cards) and the decline in price have led to a situation where sophisticated scientific calculations and modeling can be carried out on common hardware and workstations that were only possible on supercomputers a few years ago.
In the field of chemical and biological defense the use of computational calculations and modeling can significantly reduce the requirements for laboratory work involving dangerous procedures. Simulation and calculations can also help to gain a deeper understanding of molecular properties and mechanisms.
QM / DFT calculations
Quantum mechanical (QM) methods and Density Functional Theory (DFT) calculations can offer deep insights into molecular structure and reactivity. They are computationally quite “expensive” in terms of compute power (at least when meaningful parameters are selected and systems are comprised of more than just a few atoms) but offer a degree of detail that can guide experiments in the laboratory or even come up with structural and reactive parameters that make laboratory experiments unnecessary.
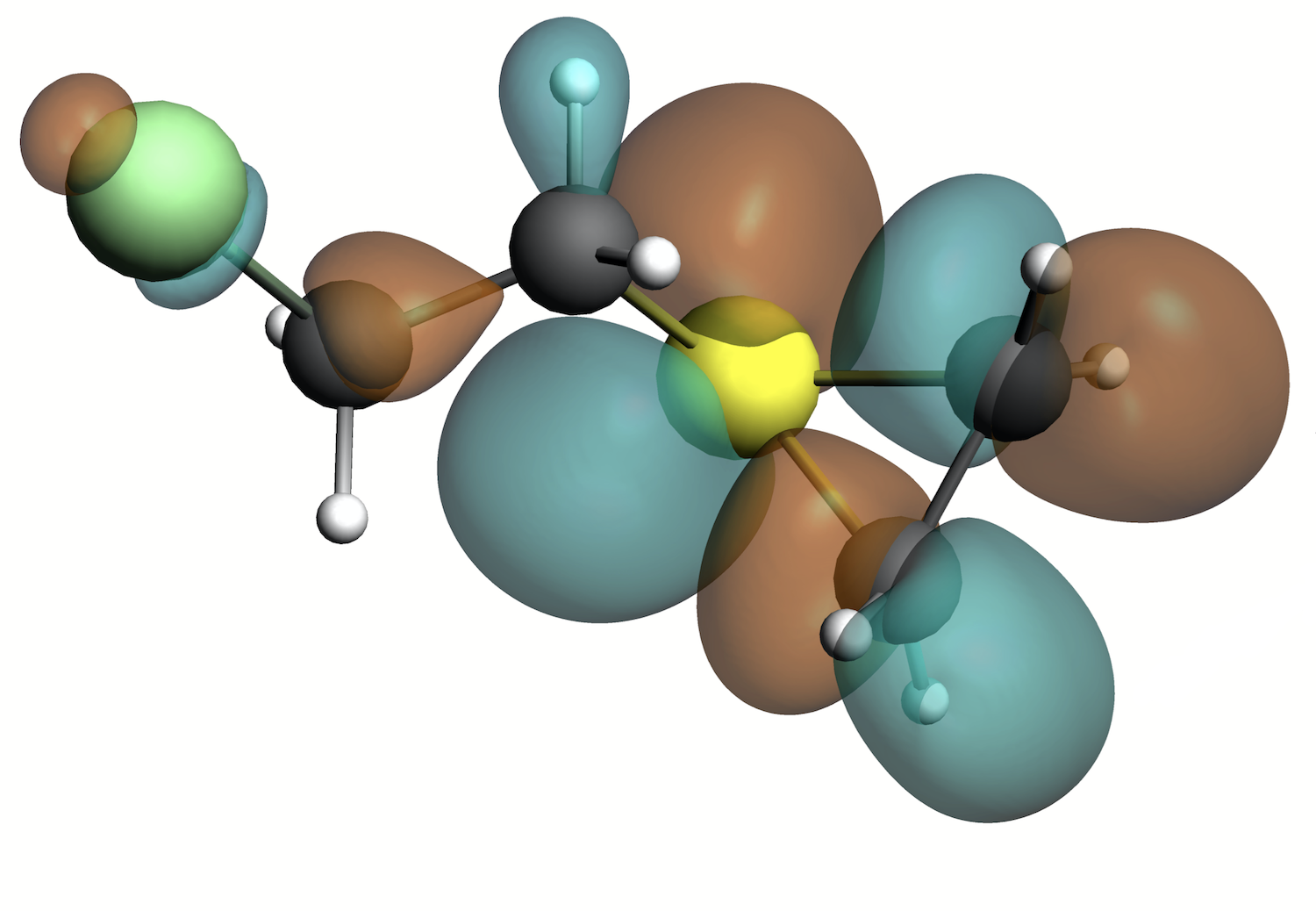
Lowest Unoccupied Molecular Orbital (LUMO) of the reactive episulfonium ion of Sulfur Mustard (HD)

Electron density of Sulfur Mustard (HD)
We work primarily with the Amsterdam Density Functional (ADF) package and modeling suite but have also employed GAUSSIAN, GAMESS and QUANTUMESPRESSO.
Molecular Dynamics (MD) simulations
Protein structures as obtain by X-ray crystallography, NMR or Cryo Electron Microscopy are important in understanding interactions with toxic compounds, the developments of antidotes and other drugs or the characterization of adducts that serve as important markers in biomedical samples. While the structures deposited in the Protein Database (PDB) are merely snapshots of a dynamic molecule the use of computational molecular dynamics simulations is a valuable technique to understand dynamic behaviors (like for example the solvent accessibility of an important part of the molecule). While simulation times were restricted to a few nanoseconds just a few years ago the vastly improved computing power available in supercomputers, cloud computing services and also smaller workstations allows longer and longer simulation times enabling insight into longer timescale phenomena. If computational demand outgrows in-house resources large MD tasks (both in terms of length and number) can be easily moved to compute resources in the cloud.
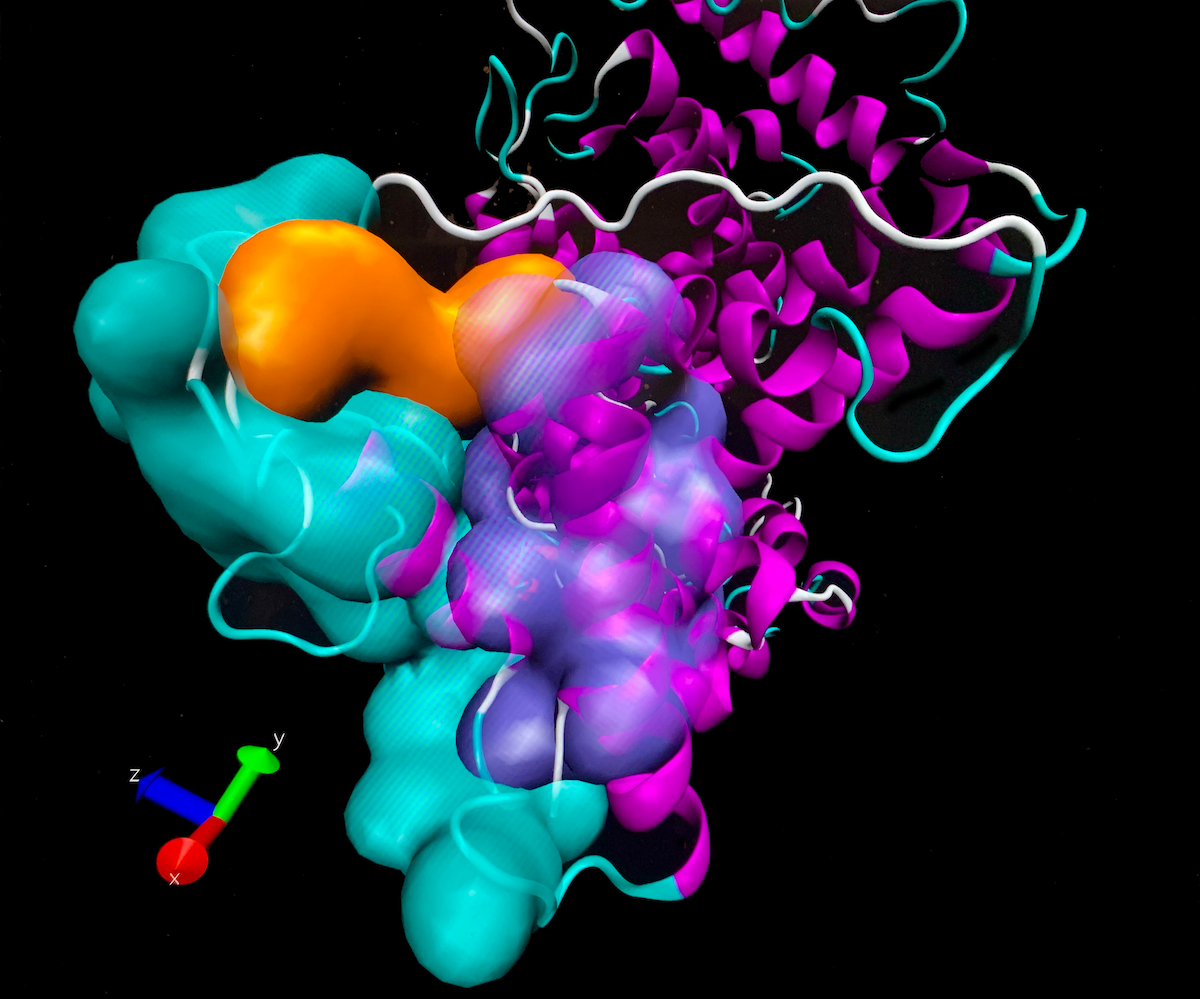
Analysis of the surrounding of a modified amino acid in Human Serum Albumin
We work primarily with GROMACS but also with NAMD and AMBER.
Structure prediction of proteins and Molecular Docking
The interaction of a small molecule – regardless if its a toxicant or a drug or antidote – with a protein target is often of prime interest. Such a protein – ligand complex can not always be characterized by structural methods and sometimes even the structure of the protein target has not been experimentally determined yet.
A major breakthrough in the area of "in-silico" protein structure prediction has been achieved recently by the introduction of the Alpha Fold and RoseTTAfold software packages, allowing the generation of computationally predicted protein structures with unprecedented accuracy. Together with the experimentally determined structures found in the Protein Database (PDB) there is now a vast amount of structural data available for scientific exploitation.
Docking is a computational method to determine the interactions of a ligand with its receptor “in-silico”. Modern software on modern hardware can derive favorite orientations of small ligands in proteins at speed suitable for high throughput computational screening. These computationally "cheap" methods can then be augmented by more sophisticated such as MD simulation, quantum mechanics/molecular mechanics (QM/MM) methods up to pure QM and DFT calculations.
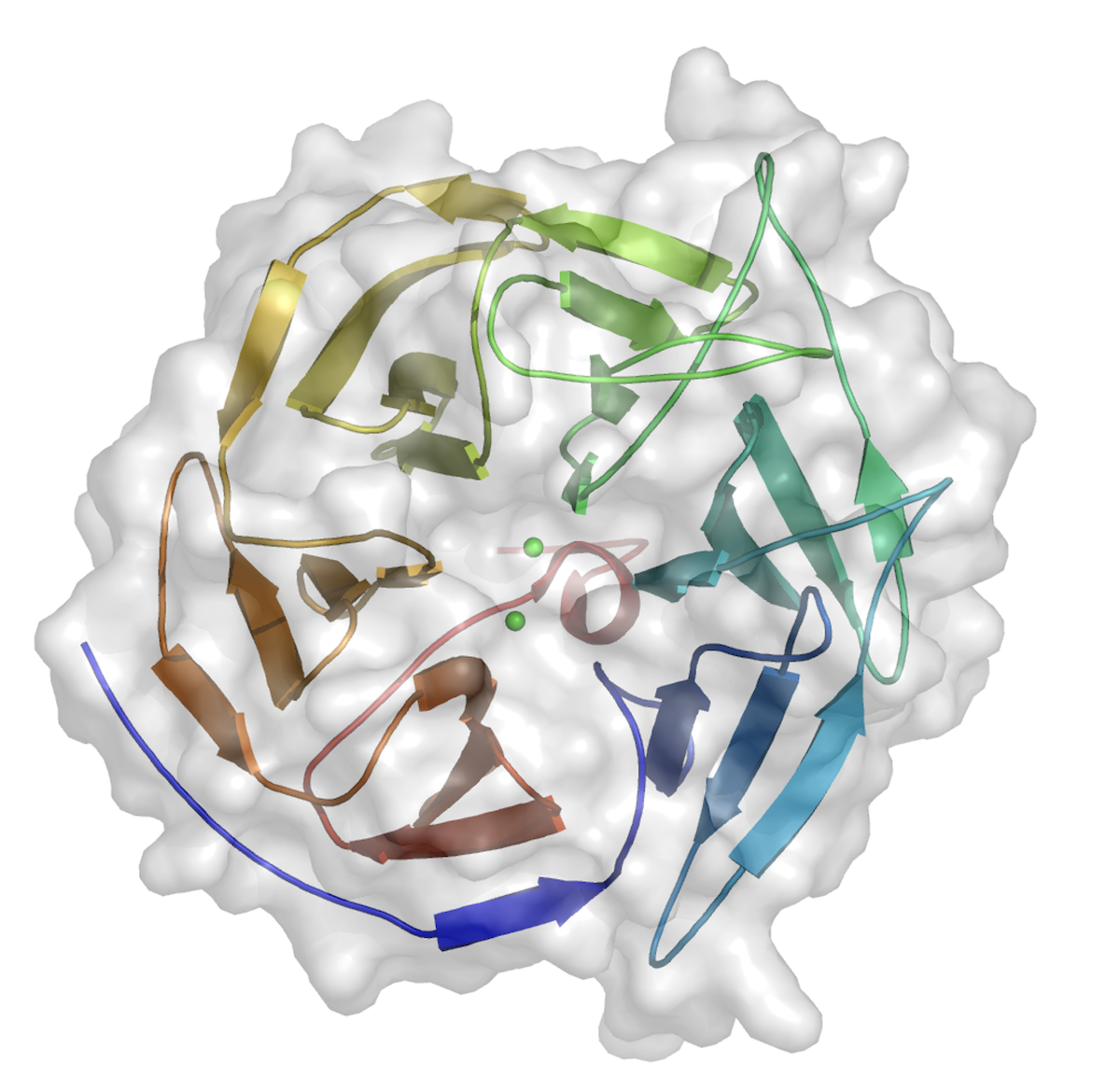
Top view of the structure of the nerve agent degrading enzyme DFPase from squid
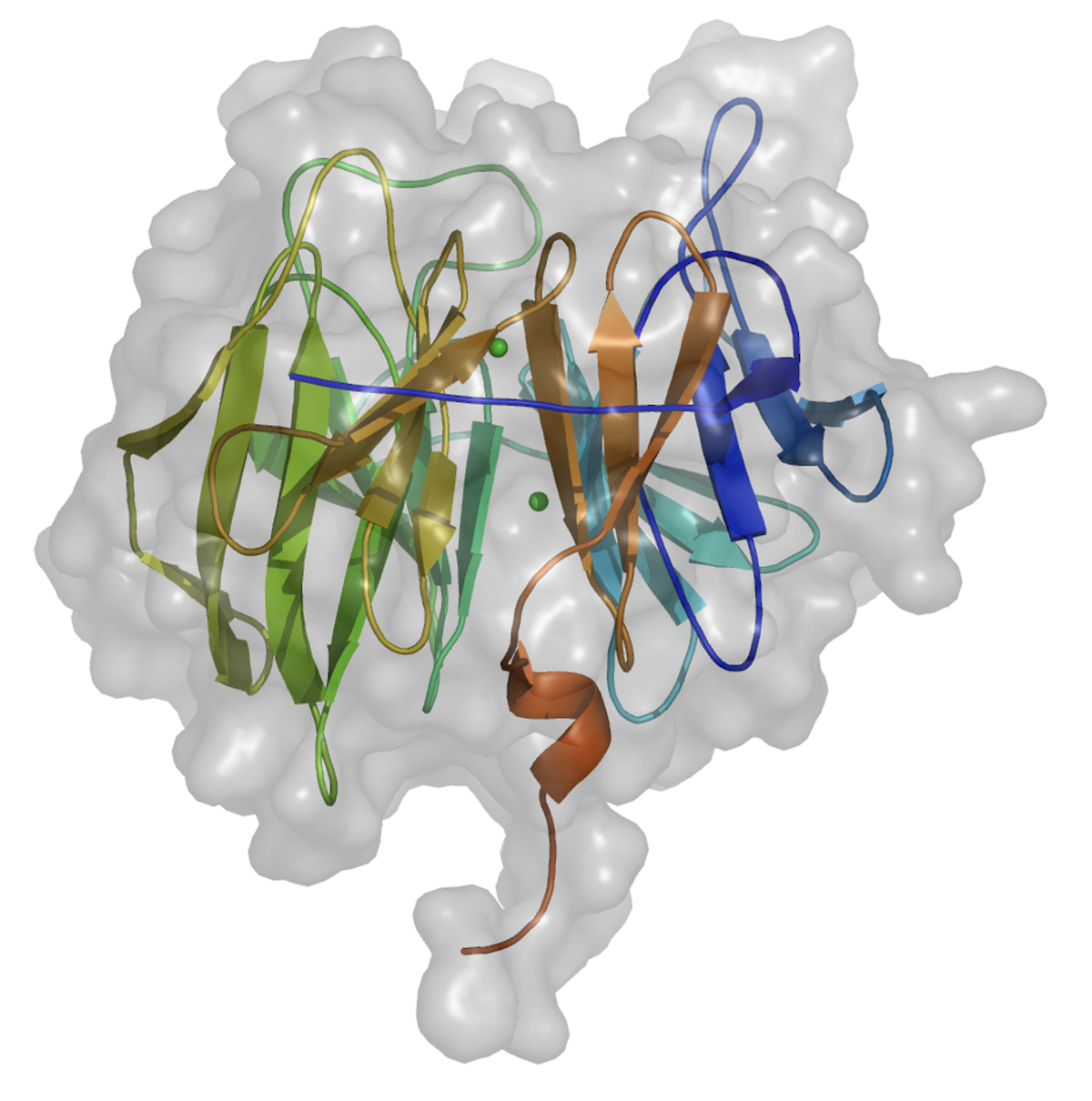
Side view of the DFPase structure
For protein structure prediction we work mainly with the Alpha Fold software package and have also use the I-TASSER software quite extensively.
For docking we work with a variety of programs including AUTODOCK (VINA), GLIDE, FlexX, HADDOCK, GOLD and others.
We can advise you on setting up computational capabilities (both in terms of hardware and software) following a detailed needs analysis.
We offer contract services in computational chemistry and molecular modeling if an in-house solution is not considered feasible.
We offer training in computational chemistry and modeling techniques based on your specific needs.